eDNA metabarcoding still never ceases to amaze, a single liter of sea water detecting hundreds of species from bread crumbs left behind. But how much do we trust our identifications of the species we have found?
Any researcher who has had the pleasure (read: nightmare) of taking tens to hundreds of millions (to now billions) of ACTGs and converting them to sequence counts of individual fish species knows that this is not an easy undertaking. And in the multilayered process of quality- control, dereplicating sequences, and assigning taxonomy there are numerous parameters that can be changed along the way (The Anacapa Toolkit has >150). And like tuning the shifters on a bicycle derailleur, changing one parameter can either cause very little change or produce dramatically different results. So besides trying every possible permutation of every parameter (which is impossible, as a practical matter) what do we do to actually test and parametrize the accuracy of our eDNA metabarcoding assignments?
Our recent paper in the California Current Large Marine Ecosystem lays the groundwork for identifying important best practices to trust species assignments. We highlight 4 best practices:
- Barcoding your species of interest makes a world of difference.
- Curated, regional reference databases outperform global/comprehensive ones.
- Confidence – Resolution Tradeoffs Abound.
- Benchmarking your marker of interest is key: one size does not fit all.
First, good high-quality reference databases are critical. You can’t even ask a single question of ecological relevance if you can’t ID a species. With our current methods we can only ID (to species level) what we have barcoded from tissues in hand – and even then, a given locus might not distinguish species from one another (like my nemesis Rockfishes). Even for extremely well studied species like fishes in California, we found that barely 40% had previously been barcoded for the MiFish Universal 12S region. A rough estimate for invertebrates in Southern California from Dean Pentcheff at the Natural History Museum of Los Angeles County is less than 30% for CO1 (so good luck doing a comprehensive benthic invert survey when 70% of species can’t be ID’d right off the bat!).
So why did eDNA work to begin with? Well there was likely a pretty solid match to sister taxa, maybe in the same family or in the same genus, but nothing great. For example, we found >17% of our millions of reads getting assigned to the family Serranidae from kelp forests in Southern California. Nice to know we have a grouper of some sort around, but not that informative especially given there are dozen species this could be matching to, some of which are threatened species and others of which are not. Knowing whether or not it’s the threatened or common species is undoubtedly important to resource managers. However, once we barcoded 605 fish species, then suddenly – BAM! We got species level ID to Kelp bass, arguably the most abundant fish in Southern California rocky reefs and one of the main recreational fisheries targets.

So clearly, making reference barcodes works. Putting in the leg work to generate high quality reference sequences pays dividends for eDNA data! (And Pro Tip – make friends with taxonomists and keep them happy since they are the true eDNA heroes). Fortunately, new methods of mitogenome skimming are now cheaper than Sanger sequencing a single gene and cover all potential regions of interest. Barcoding isn’t exactly the sexiest science, but many resource managers would argue that it makes a big difference. Being able to say “hey we found this endangered Giant Black Sea Bass in your marine protected area” vs. saying “we definitely found fish in the ocean” is the difference between decision-relevant science and getting laughed out of the room. So spend the time and effort to barcode your key target species and thank me later.
Second, curated, region-specific databases outperform global, comprehensive ones. On first hand this finding is not necessarily intuitive. You would think having all the data on hand to assign taxonomy would be the best practice, or at least I did. More data = better decisions, right? Nope!
The issue is when you have sister taxa that are not native to your region of interest. In our case this was best exemplified by the croakers. Turns out there are 6 main clades of croakers (family Sciaenidae) in Southern California (Lo et al. 2015), each of which have sister taxa across the Pacific. When you use the global comprehensive reference database (every sequence on GenBank we could get our hands on with CRUX) with species across the Pacific, you can’t get species- or even genus-level resolution of the croakers because each of these 6 clades share nearly identical 12S genes between taxa. However, by only focusing on species that we know to live in the California Current and excluding all the sister taxa from Japan and Panama that we would not realistically expect to show up in our samples, we then have the resolution to differentiate the different croakers native to California.
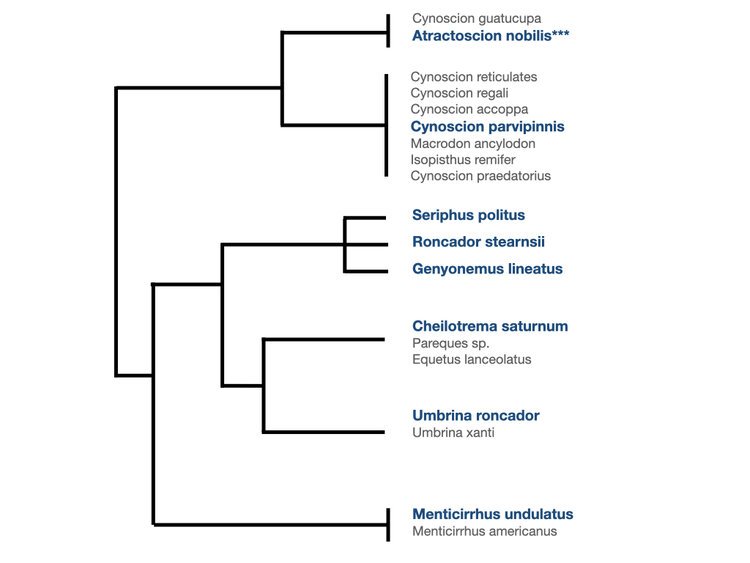
This narrower focus of course comes with drawbacks, particularly if you are concerned that one of these species is invasive or might migrate to the California Current because of climate change. But on the whole, using a curated reference database will provide the best taxonomic resolution for most applications. Obviously if you suspect an invasive species might be present then include it in your curated database. For now to triple check this, we have been assigning taxonomy to our query sequences twice, once with the regional database and again with the global database to see which sequences are being assigned differently. So far the only differences we have found have been birds and mammals… that is, species present in the global database but not in our fish specific regional database. This could of course be overcome by simply adding in all the barcodes for the marine mammals and birds in the California Current. Curating your reference library also means making sure it includes all the taxa that you expect to be getting!
Third, these findings highlight tradeoffs inherent in eDNA. On the one hand a regional database may increase accuracy for some species, but that decision has the potential unintended consequence of not allowing you to ID an invasive species that you didn’t know was in your eDNA sample. Metabarcoding is littered with these tradeoffs at every step of the process from primer selection to number of technical/sampling replicates to sequencing depth to taxonomic assignment. And often they all boil down to confidence vs. resolution in our data or in statistics – a bias-variance tradeoff.
Here we found that across taxonomic cutoff scores (we use a Bayesian lowest common ancestor classifier, and this is vaguely analogous to percent identity used by other classifiers) there is a tradeoff between accuracy of assignments and the rates of misassignments to the wrong species. This essentially raises the question – which error is worse? Is it worse to under-classify a species (e.g. be super confident but conservative in your Family level assignment when you could have arguably made the genus/species level ID with decent confidence) or is it worse to mis-classify (e.g. assign it to the wrong species when you should have kicked it back to Genus level)?
The answer – the one you probably that don’t want to hear – is “it depends” (of course). If the detection is for an endangered or invasive species and there are potentially millions of dollars of active management on the line triggered by this detection, you better damn well be confident in those detections. You do not want to be the person that misclassifies the pathogen from metabarcoding, drops $$$ to design a new drug/vaccine to target the pathogen, only to find out that whoops we misclassified that barcode and turns out it’s this other species causing the disease… (don’t be that person).
On the other hand, if the goal is to look at the ecosystem level and see how the whole community is changing in response to an environmental stressor, then you can totally afford to make some misclassification calls while increasing your overall accuracy. You might just find patterns that are only visible at the species level, but not at the family level (e.g. Rockfishes are ecologically so divergent from each other but genetically they are almost identical). And you can always cherry-pick a few species to triple check the assignment accuracy via blasting/Sanger sequencing if there are novel findings that pop out because of this. But there is no way that we can hand curate each ASV assignment; nobody’s got time for that. Still, being overall more accurate is arguably often the right strategy.
All of this came from testing just one of the 150 parameters in the Anacapa Toolkit… We can probably ignore a good 100 off the bat since they are probably minor (e.g. cutadapt trimming length), but others like Q scores apparently can make a huge difference (don’t have anything published on this, but we found that Q35 removed ~10 million reads compared to Q30 and led to dramatically increased variability across technical replicates). Clearly, it’s still the wild west out there in terms of parameters and everyone has their favorites either because they were handed down like the thermocycler profile that randomly works and you will never change or because that Tuesday morning after your fifth coffee the day before orals you said 30 sounds like a great number and never looked back. Either way, neither of these methods for conducting science is recommended. Though I truly sympathize in my soul why you don’t want to go back and question your life/PCR/parameter choices.
Which leads us to the ultimate lesson, if we have learned anything from this endeavor of trying to take DNA out of literally thin air/water is that one size does not fit all. You have to test your parameters to know what is going on under the hood in the bioinformatics pipelines. For example, the optimal taxonomic cutoff score, one that best balances confidence and resolution, for the MiFish primer (60) but this is different from the 16S V4 primers (80) and WITS primers (40). If you use the same score for bacterial 16S and throw it at fishes you will only be getting Family level assignments and you will have a much less exciting day than the one where you say: “WOAH I found blue whale in this bottle of water off Santa Cruz Island!”. All of this is to say that taking an optimal parameter from one primer set and throwing it to another is not the way we should be conducting metabarcoding. The 97% taxonomic identity comes from an average of differences in mtDNA between vertebrates. It is truly an abstract average number. We know that this is not even true for all vertebrates (Rockfishes, for example). Does it make sense to apply to crustaceans? Who the hell knows?! But if you don’t test it then you don’t know. And if you don’t know then we are just the blind leading the blind down the mine field of rabbit holes that is metabarcoding taxonomic assignment. If I learned anything from the 90s is that this is a bad way to play Mine Sweeper. You might randomly win, but 99.999% of the time you are going to have a bad day.
So where does that leave us?
We need to be benchmarking each primer set and evaluating the optimal parameter selection for each. Yes, it sounds awfully tedious, but it’s clearly warranted by the data and we have the tools do this (and are working on making them even easier to implement – look out for rCRUX). Robert Edgar has a brilliant set of scripts to run taxonomy by cross validation (TAXXI) to determine how changing a single parameter, reference database, or entire pipeline effects taxonomic assignment scores. The tools are out there and it just takes some legwork to find the optimal settings.
It’s worth checking every piece of your bike to know how it works – changing the large gears has a lot more impact than changing a small gear. Likewise changing the angle of the derailleur hanger can have a huge impact on shifting performance while changing the tension in your barrel adjusters is minor. The only way we are going to climb up this hill of eDNA metabarcoding and be able to effectively and accurately assign taxonomy is to know which parameters are large gears, which are small, and which are barrel adjusters. To do this we are going to have to take our pipelines apart and systematically fine tune the parameters to know which ones matter and which ones not so much. Sure you can borrow someone else’s bike and jump on and try to climb to the top, but I promise you the ride is a lot more enjoyable in low gear. That being said, if your barrel adjuster is off for the ride will you even notice? If you get to the top does it even matter? But for the love of your fellow eDNA scientists and humanity as a whole if you do find the low gear, please, tell your friends. Scream it from the top of mount eDNA. We will buy you multiple cases of beer for saving us from the long arduous slog of parsing sequences so we can get to the “Woah I found Great White Shark off of Palos Verdes” moments a lot faster.